Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
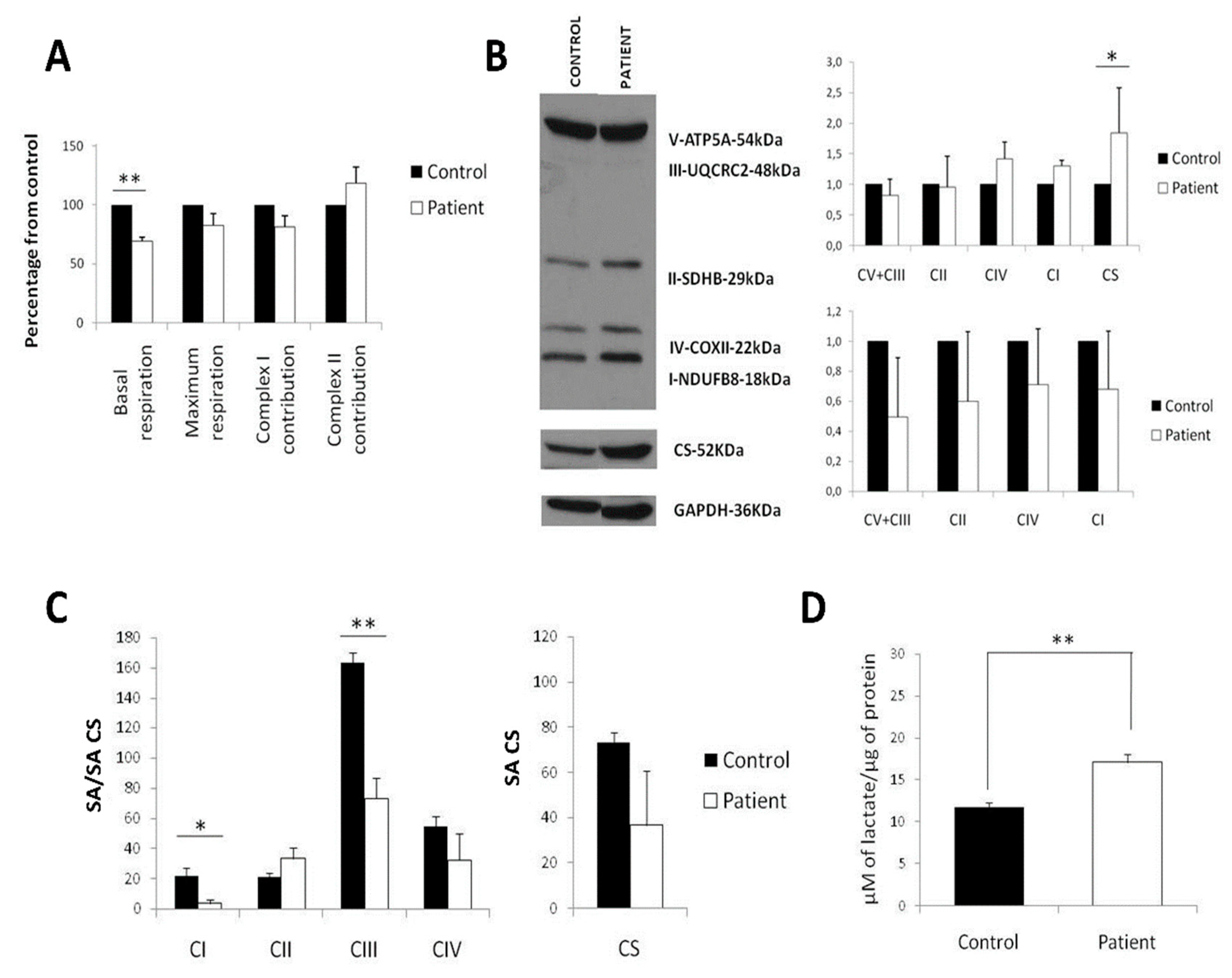
2.1. Diminished Respiration in LS Fibroblasts Caused by a Decrease of Mitochondrial Mass
2.2. Generation of the Control iPSc Line N44SV.1
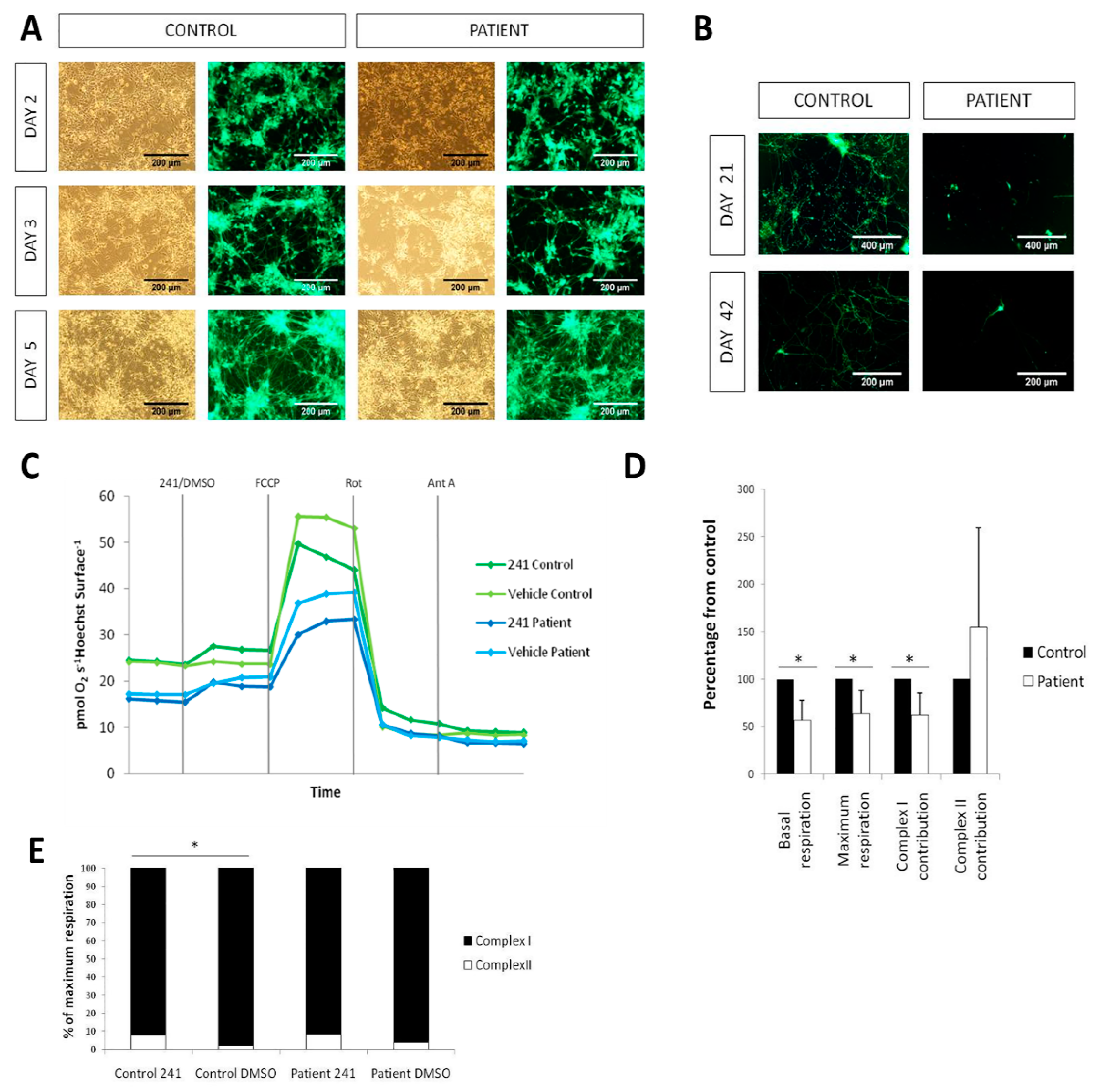
2.3. LS iPSCs Manifest a Decreased Basal Respiration and a Combined RC Deficiency
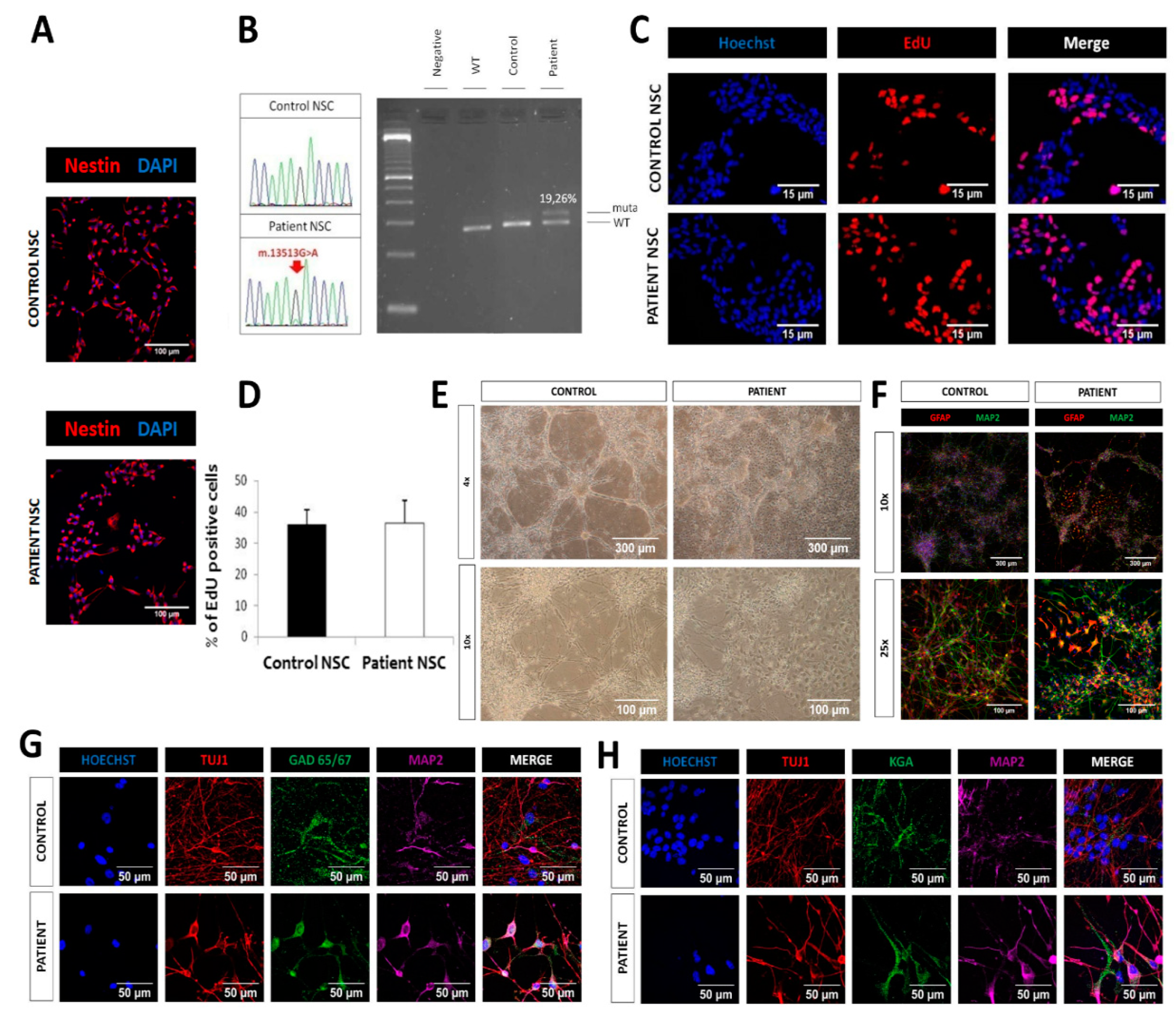
2.4. Similar Proliferation and Differentiation Capacity of LS iPSC-Derived NSCs
2.5. Cell death and Complex I deficiency in LS iPSC-Derived Neurons
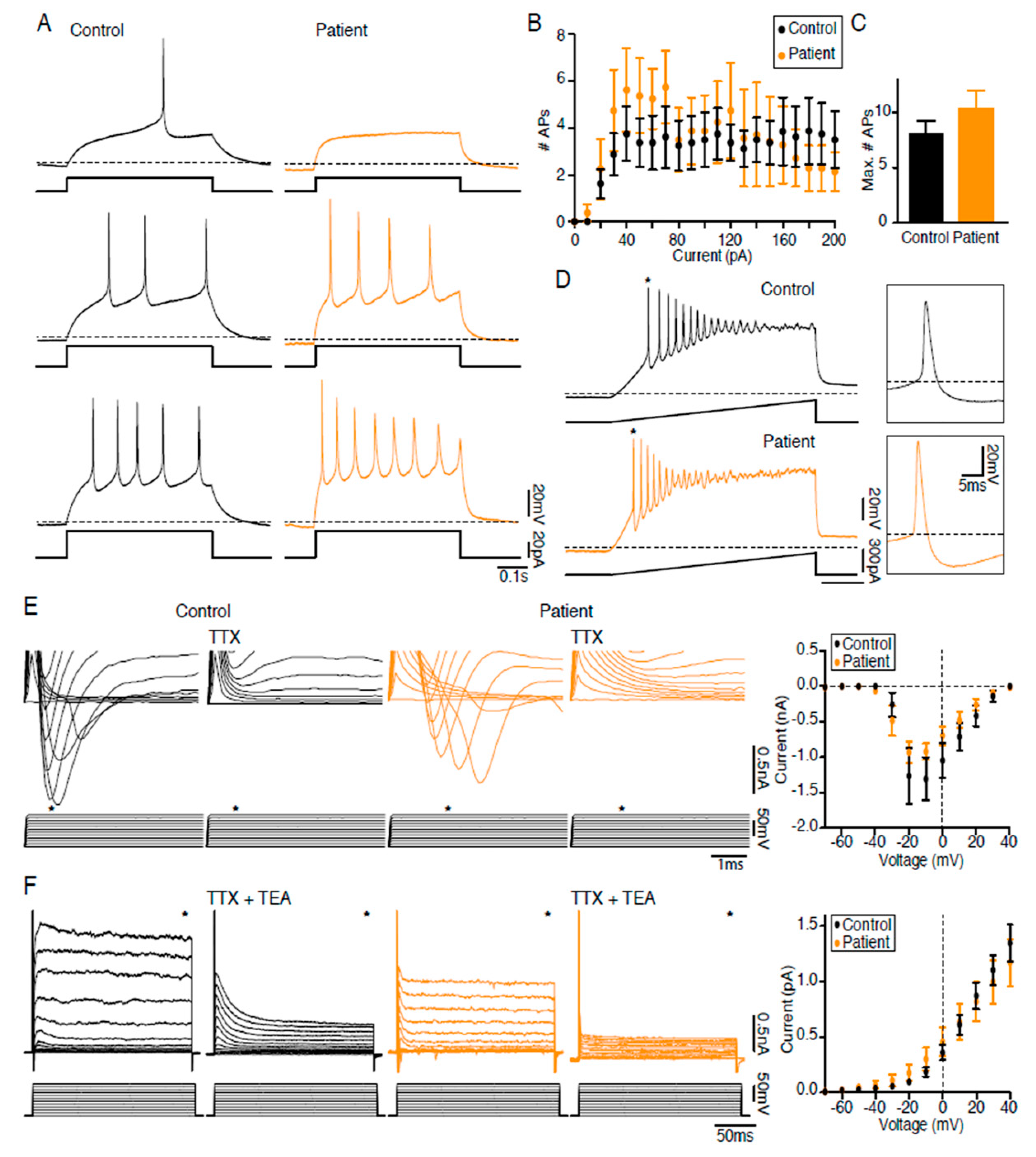
2.6. LS iPSC-Derived Neurons are Functional
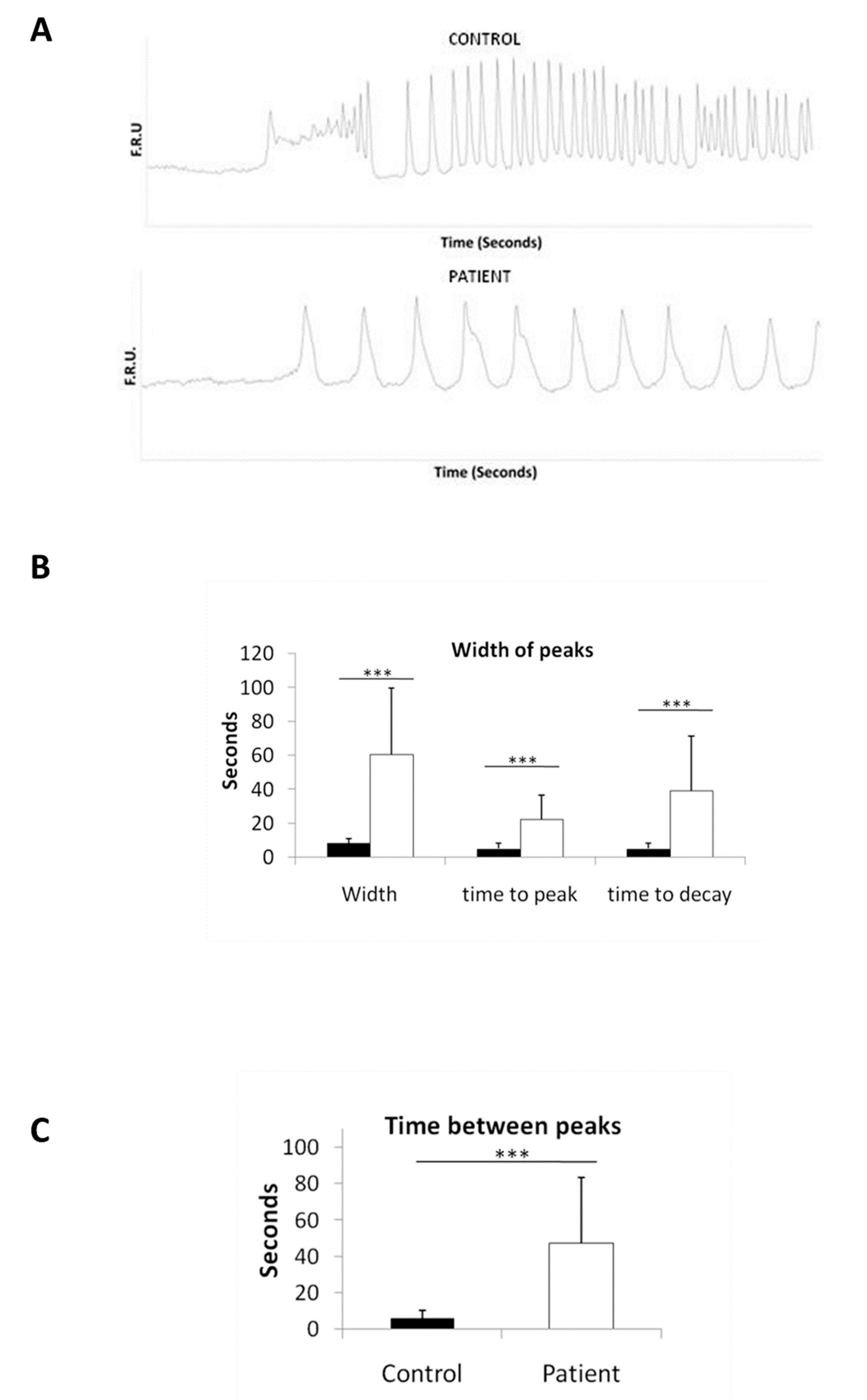
2.7. Disturbed Calcium Regulation in LS iPSC-Derived Neurons
3. Discussion
4. Materials and Methods
4.1. Cell Culture, iPSC Generation and Characterization
4.2. Oxygen Consumption
4.3. Protein Extraction and Western Blot
4.4. Lactic Acid Production
4.5. Respiratory Chain Activity Determination by Spectrophotometry
4.6. NSC Generation and Neuronal Differentiation
4.7. Mutation Analysis and Heteroplasmy Quantification of mtDNA m.13513G>A Mutation
4.8. Proliferation Assay
4.9. Immunofluorescence
4.10. iN Morphology Analysis
4.11. Electrophysiology
4.12. Calcium Imaging
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LS | Leigh syndrome |
| MD | Mitochondrial disorder |
| iPSCs | Induced pluripotent stem cells |
| mtDNA | Mitochondrial DNA |
| NSCs | Neural stem cells |
| OXPHOS | Mitochondrial oxidative phosphorylation system |
| DMSO | Dimethyl sulfoxide |
| FCCP | Carbonyl cyanide p-trifluoromethoxyphenylhydrazone |
| RC | Respiratory chain |
| ROS | Reactive oxygen species |
| MELAS | Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke |
| ATP | Adenosine triphosphate |
| ETC | Electron transfer chain |
| rRNA | Ribosomal ribonucleic acid |
| tRNA | Transfer RNA |
| CS | Citrate synthase |
| ∆Ψmit | Mitochondrial membrane potential |
| AP | Action potential |
| FRU | Fluorescence relative units |
References
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, A.; Zanella, S.; Koch, H.; Kruse, S.E.; Lee, D.; Ramirez, J.M.; Palmiter, R.D. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Investig. 2012, 122, 2359–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochim. Biophys. Acta 2015, 1847, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Mlody, B.; Lorenz, C.; Inak, G.; Prigione, A. Energy metabolism in neuronal/glial induction and in iPSC models of brain disorders. Semin. Cell Dev. Biol. 2016, 52, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Monlleo-Neila, L.; Toro, M.D.; Bornstein, B.; Garcia-Arumi, E.; Sarrias, A.; Roig-Quilis, M.; Munell, F. Leigh Syndrome and the Mitochondrial m.13513G>A Mutation: Expanding the Clinical Spectrum. J. Child Neurol. 2013, 28, 1531–1534. [Google Scholar] [CrossRef] [Green Version]
- Galera, T.; Zurita-Diaz, F.; Garesse, R.; Gallardo, M.E. iPSCs, a Future Tool for Therapeutic Intervention in Mitochondrial Disorders: Pros and Cons. J. Cell. Physiol. 2016, 231, 2317–2318. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Folmes, C.D.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5, e13378. [Google Scholar] [CrossRef] [PubMed]
- Galera, T.; Zurita, F.; Gonzalez-Paramos, C.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernandez, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with Leigh syndrome. Stem Cell Res. 2016, 16, 63–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [Green Version]
- Ehinger, J.K.; Piel, S.; Ford, R.; Karlsson, M.; Sjovall, F.; Frostner, E.A.; Morota, S.; Taylor, R.W.; Turnbull, D.M.; Cornell, C.; et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016, 7, 12317. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Hernandez, A.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gomez Izquierdo, L.; Cotan, D.; Navas, P.; Sanchez-Alcazar, J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Cotan, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodriguez-Hernandez, A.; Gomez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2669–2687. [Google Scholar]
- De la Mata, M.; Garrido-Maraver, J.; Cotan, D.; Cordero, M.D.; Oropesa-Avila, M.; Izquierdo, L.G.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; Ybot, P.; et al. Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurother. J. Am. Soc. Exp. NeuroTher. 2012, 9, 446–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; Giorgio, V.; Cali, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016, 23, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.; Delmiro, A.; Blazquez, A.; Ugalde, C.; Arenas, J.; Martin, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef]
- Prigione, A.; Hossini, A.M.; Lichtner, B.; Serin, A.; Fauler, B.; Megges, M.; Lurz, R.; Lehrach, H.; Makrantonaki, E.; Zouboulis, C.C.; et al. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS ONE 2011, 6, e27352. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [Green Version]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnle, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.; Kekesi, O.; Morley, J.W.; Tapson, J.; Breen, P.P.; van Schaik, A.; Buskila, Y. Calcium Imaging of AM Dyes Following Prolonged Incubation in Acute Neuronal Tissue. PLoS ONE 2016, 11, e0155468. [Google Scholar] [CrossRef]
- Duchen, M.R. Contributions of mitochondria to animal physiology: From homeostatic sensor to calcium signalling and cell death. J. Physiol. 1999, 516 Pt 1, 1–17. [Google Scholar] [CrossRef]
- Collins, T.J.; Lipp, P.; Berridge, M.J.; Li, W.; Bootman, M.D. Inositol 1,4,5-trisphosphate-induced Ca2+ release is inhibited by mitochondrial depolarization. Biochem. J. 2000, 347, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Aure, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Sahara, N.; Kanaan, N.M.; Tsukita, K.; Kondo, T.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Kawakami, K.; Hotta, A.; et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016, 6, 34904. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, A.; Janke, J.; Doring, H.J.; Leder, O. Myocardial fiber necrosis due to intracellular Ca overload-a new principle in cardiac pathophysiology. Recent Adv. Stud. Card. Struct. Metab. 1974, 4, 563–580. [Google Scholar]
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Arnao, V.; Renda, C.; Pinto, A.; Licata, G. Neuron protection as a therapeutic target in acute ischemic stroke. Curr. Top. Med. Chem. 2009, 9, 1317–1334. [Google Scholar] [CrossRef]
- McKenzie, M.; Duchen, M.R. Impaired Cellular Bioenergetics Causes Mitochondrial Calcium Handling Defects in MT-ND5 Mutant Cybrids. PLoS ONE 2016, 11, e0154371. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Diaz, F.; Garesse, R.; Gallardo, M.E. The mutation m.13513G>A impairs cardiac function, favoring a neuroectoderm commitment, in a mutant-load dependent way. J. Cell. Physiol. 2019, 234, 19511–19522. [Google Scholar] [CrossRef] [PubMed]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; Mousson de Camaret, B.; Slama, A.; Lunardi, J.; Mazat, J.P.; Lombes, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.G.; Tornero, D.; Canals, I.; Ahlenius, H.; Kokaia, Z. In Vitro Functional Characterization of Human Neurons and Astrocytes Using Calcium Imaging and Electrophysiology. Methods Mol. Biol. 2019, 1919, 73–88. [Google Scholar] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galera-Monge, T.; Zurita-Díaz, F.; Canals, I.; Grønning Hansen, M.; Rufián-Vázquez, L.; Ehinger, J.K.; Elmér, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. Int. J. Mol. Sci. 2020, 21, 3191. https://doi.org/10.3390/ijms21093191
Galera-Monge T, Zurita-Díaz F, Canals I, Grønning Hansen M, Rufián-Vázquez L, Ehinger JK, Elmér E, Martin MA, Garesse R, Ahlenius H, et al. Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. International Journal of Molecular Sciences. 2020; 21(9):3191. https://doi.org/10.3390/ijms21093191
Chicago/Turabian StyleGalera-Monge, Teresa, Francisco Zurita-Díaz, Isaac Canals, Marita Grønning Hansen, Laura Rufián-Vázquez, Johannes K. Ehinger, Eskil Elmér, Miguel A. Martin, Rafael Garesse, Henrik Ahlenius, and et al. 2020. "Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons" International Journal of Molecular Sciences 21, no. 9: 3191. https://doi.org/10.3390/ijms21093191